

Introduction
What if, a small sample of each baby’s saliva was sent out to a lab, where—for just a few dollars—the baby’s DNA was analyzed and a multitude of “risk scores” returned? These would not be diagnoses but instead, prognostication: This baby is at elevated risk for developing heart disease in 40 years, Is more likely than average to suffer from depression or schizophrenia someday, and how to deal?. This baby may have a very high IQ or a low one.“Blueprint: How DNA Makes Us Who We Are, Book”, he makes the case that in the very near future, we will be able to know, at birth, something about our risk for developing nearly every imaginable psychological and
illness—our “risk scores”—and that this knowledge will help researchers develop new treatments and interventions, and will help all of us live lives better in keeping with our individual natures.
What’s DNA (Deoxyribonucleic Acid)?
Carries genetic instructions in all living things, It’s like the description of all information about your body like your eyes color, skin color, genetic diseases (like sugar disease), And what’s the responsible gene?
Before diving into DNA description let’s descript where to find DNA?
Cells are the smallest units of life, that contain nuclei, A Nucleus contains 23 pairs of chromosomes, A chromosome is a long string of DNA molecule with part or all of the genetic material of an organism,

DNA stands for deoxyribonucleic acid, Each string of DNA looks like a twisted ladder. Scientists call this a double helix. DNA is like a code containing all the instructions that tell a cell what to do, It is made up of genes, Humans have around 25,000 genes in total.

You inherit half your DNA from your mother and a half from your father, So you have 2 copies of every gene. Your genes carry all the information that makes you.
For example,
they tell your body to have blonde hair or brown skin, or green eyes, and they tell your cells:
– What sort of cell to be?
– How to behave?
– When to grow and reproduce?
– When to die?
“Some genes control how much each cell grows and divides”.
DNA Shape: is double-helix “consists of two strands that wind around one another to form a shape double helix”.DNA consist of:
1. Sugar (deoxyribose)
2. Phosphate
3. Base pairs
Sugar & Phosphate groups called “sugar-phosphate backbone”
Base pairs made of four types of nitrogen bases: Adenine (A), Thymine (T), Guanine (G), and Cytosine. We always call them A, C, G, T.
“Base pairs consist of four chemicals linked together via hydrogen bonds in any possible order making a chain, which gives one thread of the DNA double-helix. And the second thread of the double-helix balance the first. So if you have ‘A’ on the first thread, you have to have ‘T’ on the second & if you have ‘C’ on the first thread, you have to have ‘G’ on the second, A and T always balance each other & C and G balance each other, So once you identify one thread of the helix, you can always spell the other”.

- Attached to each sugar is one of four bases: adenine (A), cytosine (C), guanine (G), and thymine (T).
- Phosphate groups attached with Sugar groups bond called “sugar-phosphate backbone”.
- Phosphate with Suger & 1/2 Base pair called “Nucleotide”.

The order, or sequence, of these bases, determines what biological instructions are contained in a strand of DNA. For example, the sequence ATCGTT might instruct for blue eyes, while ATCGCT might instruct for brown.

A human genome has about 6 billion characters or letters. If you think the genome(the complete DNA sequence) is like a book, it is a book about 6 billion letters of “A”, “C”, “G” and “T”. Scientists find most parts of the human genomes are like each other.
Machine Learning Algorithm
In the past few decades, we have witnessed the revolutionary development of biomedical research and biotechnology and the explosive growth of biomedical data. The problem has changed from the accumulation of biomedical data to how to mine useful knowledge from the data. On the one hand, the rapid development of biotechnology and biological data analysis methods has led to the emergence of a challenging new field, bioinformatics. On the other hand, the continuous development of biological data mining technology has produced a large number of effective and well-scalable algorithms. How to build a bridge between the two fields of machine learning and bioinformatics to successfully analyze biomedical data is worthy of attention and research. In particular, we should analyze how to use data mining for effective biomedical data analysis, and outline some research questions that may stimulate the further development of powerful biological machine learning algorithms.
So in this article, we will understand how to interpret a DNA structure and learn machine learning algorithms to classify each DNA sequence according to its Gene family and use it to build a prediction model on DNA sequence data.
Basic Process of Machine learning project:
The Machine-Learning process is a discipline that combines classic statistical tools with computer science algorithms. This discipline aims to mine knowledge from large amounts of data for scientific, computational, or industrial use. we comprehensively describe the process of machine-learning from six aspects.
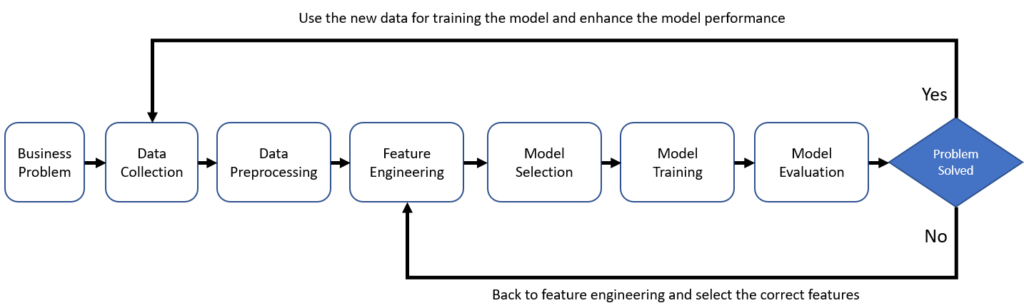
- Identify Business Problem: Understand very well the business problem you are trying to solve and what is the expected outcome of this problem
- Data Collection: Collect all required data from the different source systems related to your problem.
- Data Preprocessing: is the process of cleaning and transforming raw data prior to processing and analysis. It is an important step prior to processing and often involves reformatting data, making corrections to data, and combining data sets to enrich data.
– Data Cleaning: data sets often have missing data and inconsistent data, low data quality will
have a serious negative impact on the information extraction process, Therefore,
deleting incomplete, or inconsistent data is the first step in data mining.
– Data Integration: If the source of the data to be studied is different, it must be aggregated
consistently. - Feature Engineering: this includes all changes to the data from once it has been cleaned up to when it is ingested into the machine learning model. In this step you select and prepare the features that will be used in your machine learning model and you make sure it is in the proper format as expected by the Machine Learning Model.
- Model Selection: Select the appropriate model according to the problem and make subsequent improvements.
- Training & Tuning of the Model: The process of training an ML model involves providing an ML algorithm (that is, the learning algorithm) with training, data to learn from. The term ML model refers to the model artifact that is created by the training process.
- Model Evaluation: This is a systematic approach that will guide in measuring the efficiency and effectiveness of training.
- Feedback: In this feedback loop we check the model performance against the problem we are trying to solve, and we can use the output of the model to enhance the model training based on live data
Getting Dataset (Data selection):
I offer 3 insights categories of data sets:
- Human dataset: Represent DNA sequences of humans that contain 7 Gene classes.
- Chimpanzee dataset: Represent DNA sequences of chimpanzees that contain 7 Gene classes.
- Dog dataset: Represent DNA sequences of dogs that contain 7 Gene classes.
Let’s Describe Dataset:
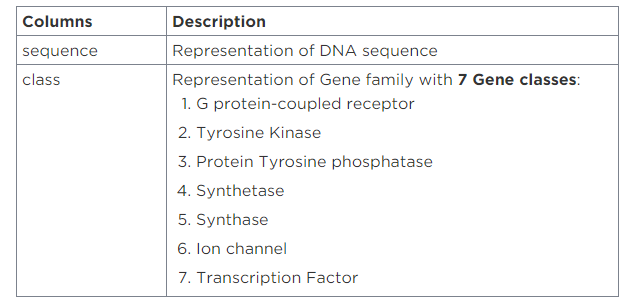

Let’s Descript each Gene & What’s important? :
In this project, We selected 7 Genes:
1. G protein-coupled receptors (GPCRs): form a large group of evolutionarily related proteins that are cell surface receptors that detect molecules outside the cell and activate cellular responses, The ligands that bind and activate these receptors include light-sensitive compounds, odors, pheromones, hormones, and neurotransmitters, and vary in size from small molecules to peptides to large proteins. G protein-coupled receptors are involved in many diseases (like Cancer disease).
2. Tyrosine kinase: are important mediators of the signaling cascade, determining key roles in diverse biological processes like growth, differentiation, metabolism, and apoptosis in response to external and internal stimuli. Recent advances have implicated the role of tyrosine kinases in the pathophysiology of cancer. Though their activity is tightly regulated in normal cells, they may acquire transforming functions due to mutation(s), overexpression, and autocrine paracrine stimulation, leading to malignancy. Constitutive oncogenic activation in cancer cells can be blocked by selective tyrosine kinase inhibitors and thus considered a promising approach for innovative genome-based therapeutics.
3. Protein tyrosine phosphatases: are a group of enzymes that remove phosphate groups from phosphorylated tyrosine residues on proteins.
4. Protein tyrosine phosphatases (PTPs): have been recognized as the main targets for several diseases, including cancer, and great efforts have been made to identify novel PTPs inhibitors to fight cancer progression and metastasis formation. Here, we summarize recent evidence underlining the efficacy of this strategy for melanoma treatment. In particular, we illustrate how this approach could be applied to target both cancer cells and the immune infiltrate of tumors, providing a new promising adjuvant therapy for the treatment of melanoma.
5. Aminoacyl-tRNA synthetases (AARSs): play a fundamental role in protein translation, linking transfer RNAs to their cognate amino acids. But in the hundreds of millions of years that they’ve existed, these synthetases (AARSs) have picked up several side jobs. One of these is to manage the development of vertebrate vasculature.
6. Ion channels: allow ions to pass through the channel pore. Their functions include establishing a resting membrane potential, shaping action potentials and other electrical signals by gating the flow of ions across the cell membrane, controlling the flow of ions across secretory and epithelial cells, and regulating cell volume. Ion channels are present in the membranes of all cells. Ion channels are one of the two classes of iontophoretic proteins, the other being ion transporters.
7. Transcription Factor: are proteins that regulate the transcription of genes—that is, their copying into RNA, on the way to making a protein. The human body contains many transcription factors. So does the body of a bird, tree, or fungus! Transcription factors help ensure that the right genes are expressed in the right cells of the body, at the right time.
2- Data Processing
import os
#get all files in dataset directory
for dirname, _, filenames in os.walk('.dataset'):
for filename in filenames:
print(os.path.join(dirname, filename))
import pandas as pd
#read dataset files and put it in dataframes
human_dna = pd.read_table('./dataset/human.txt')
chimpanzee_dna = pd.read_table('./dataset/chimpanzee.txt')
dog_dna = pd.read_table('./dataset/dog.txt')
#show first 5 rows in human dataset
human_dna.head()
Let’s display how many DNA sequences in each class?
1. Human Dataset
#display how many DNA sequences in each class?
human_dna['class'].value_counts().sort_index().plot.bar()
plt.title("Class distribution of Human NA")
2. Chimpanzee Dataset
#display how many DNA sequences in each class?
chimpanzee_dna['class'].value_counts().sort_index().plot.bar()
plt.title("Class distribution of Chimpanzee DNA")
3. Dog Dataset
#despaly how many DNA sequence in each class?
dog_dna['class'].value_counts().sort_index().plot.bar()
plt.title("Class distribution of Dog DNA")
Data Cleaning
- Change all sequence characters to small letters.
- Check if DNA includes any character not in (A, C, G, T) put it as (z) char.
#lets descripe one dna sequence of human
one_dna = np.array(human_dna.head(1))
one_dna = str(one_dna).split(' ')
seq = one_dna[0].replace("[['",'')
seq = seq.replace("'",'')
print(seq)
#create list of lovercase char and check if any char not in (a, c, g, t) put it as (z) char
def string_to_array(seq_string):
seq_string = seq_string.lower()
seq_string = re.sub('[^acgt]', 'z', seq_string)
seq_string = np.array(list(seq_string))
return seq_string
string_to_array(seq)
Feature Selection
As show sequence DNA is characters but Machine learning models require numerical values or feature matrices. So we can encode these characters into metrics
Let’s describe 3 general approaches to encode sequence DNA:
1- Ordinal encoding DNA Sequence
2- One-hot encoding DNA Sequence
3- K-mer counting: the language of DNA sequence
1- Ordinal encoding DNA Sequence
In this approach, we need to encode each nitrogen bases as an ordinal value. For example “A, T, G, C” becomes [0.25, 0.5, 0.75, 1.0]. Any other base such as “Z” can be a 0.
def ordinal_encoder(my_array):
label_encoder = LabelEncoder()
label_encoder.fit(np.array(['a','c','g','t','z']))
integer_encoded = label_encoder.transform(my_array)
#print(integer_encoded)
float_encoded = integer_encoded.astype(float)
float_encoded[float_encoded == 0] = 0.25 # A
float_encoded[float_encoded == 1] = 0.50 # C
float_encoded[float_encoded == 2] = 0.75 # G
float_encoded[float_encoded == 3] = 1.00 # T
float_encoded[float_encoded == 4] = 0.00 # anything else, lets say z
return float_encoded
#Let’s try it out a simple short sequence:
seq_test = 'attcgxffgtg'
ordinal_encoder(string_to_array(seq_test))
2- One-hot encoding DNA Sequence
“A,C,G,T,Z” would become [1,0,0,0,0], [0,1,0,0,0], [0,0,1,0,0], [0,0,0,1,0], [0,0,0,0,1], and these one-hot encoded vectors can either be concatenated or turned into 2-dimensional
arrays
def one_hot_encoder(seq_string):
label_encoder = LabelEncoder()
label_encoder.fit(np.array(['a','c','g','t','z']))
int_encoded = label_encoder.transform(seq_string)
onehot_encoder = OneHotEncoder(sparse=False, dtype=int)
int_encoded = int_encoded.reshape(len(int_encoded), 1)
onehot_encoded = onehot_encoder.fit_transform(int_encoded)
return onehot_encoded
#So let’s try it out with a simple short sequence:
seq_test = 'attcgxffgtg'
one_hot_encoder(string_to_array(seq_test))
Note:
“None of these above methods results in vectors of uniform length, and that is a necessity for feeding
data to a classification or, regression algorithm. So with the above methods, you have to resort to things
like truncating sequences or padding with “n” or “0” to get vectors of uniform length”
3- K-mer counting DNA language:
DNA and protein sequences can be seen as the language of life. The language encodes instructions as well as functions for the molecules that are found in all life forms. The sequence language resemblance continues with the genome as the book, subsequences (genes and gene families) are sentences and chapters, k-mers and peptides are words, nucleotide bases and amino acids are the alphabets.
Let’s describe how it works:
The method we use here is manageable and easy. We first take the long biological sequence and break it down into k-mer length
overlapping “words”. For example, if we use “words” of length 6 (hexamers), “ATGCATGCA” becomes: ‘ATGCAT’, ‘TGCATG’, ‘GCATGC’,
‘CATGCA’. Hence our example sequence is broken down into 4 hexamer words.
def Kmers_funct(seq, size=6):
return [seq[x:x+size].lower() for x in range(len(seq) - size + 1)]
#So let’s try it out with a simple sequence:
mySeq = 'GTGCCCAGGTTCAGTGAGTGACACAGGCAG'
#size of each sub-seq = 6
words = Kmers_funct(mySeq, size=6)
print(words)
#change words to sentences by concatenates all words
joined_sentence = ' '.join(words)
joined_sentence
In This project I will use K-mer counting DNA language:
- Apply k-mers function to all datasets & join all words to the list
- Then Add “words” column & drop “sequence” column
#apply kmers function to all datasets & join all words to list
#then add words column & drop sequence column
human_dna['words'] = human_dna.apply(lambda x: Kmers_funct(x['sequence']), axis=1)
human_dna = human_dna.drop('sequence', axis=1)
chimpanzee_dna['words'] = chimpanzee_dna.apply(lambda x: Kmers_funct(x['sequence']), axis=1)
chimpanzee_dna = chimpanzee_dna.drop('sequence', axis=1)
dog_dna['words'] = dog_dna.apply(lambda x: Kmers_funct(x['sequence']), axis=1)
dog_dna = dog_dna.drop('sequence', axis=1)
#show first 5 rows in human dataset
human_dna.head()
Next Step: We need to now convert the lists of k-mers for each gene into string sentences of words that can be used to create the Bag of Words model. We will make a target variable y to hold the class labels.
#apply to human
human_texts = list(human_dna['words'])
for item in range(len(human_texts)):
human_texts[item] = ' '.join(human_texts[item])
#separate labels
y_human = human_dna.iloc[:, 0].values # y_human for human_dna
#apply to chimpanzee
chimpanzee_texts = list(chimpanzee_dna['words'])
for item in range(len(chimpanzee_texts)):
chimpanzee_texts[item] = ' '.join(chimpanzee_texts[item])
#separate labels
y_chim = chimpanzee_dna.iloc[:, 0].values # y_chim for chimp_dna
#apply to dog
dog_texts = list(dog_dna['words'])
for item in range(len(dog_texts)):
dog_texts[item] = ' '.join(dog_texts[item])
#separate labels
y_dog = dog_dna.iloc[:, 0].values # y_dog for dog_dna
#show Human class labels as example
print(y_human)
So, for humans we have 4380 genes converted into uniform length feature vectors of 4-gram k-mer (length 6) counts. For chimp and dog, we have the same number of features with 1682 and 820 genes respectively.
cv = CountVectorizer(ngram_range=(4,4)) #The n-gram size of 4 is previously determined by testing
X_human = cv.fit_transform(human_texts)
X_chimpanzee = cv.transform(chimpanzee_texts)
X_dog = cv.transform(dog_texts)
print(X_human.shape)
print(X_chimpanzee.shape)
print(X_dog.shape)
Once the data is divided into inputs and labels, the final preprocessing step is to divide data into training and test sets. Luckily, the “model_selection” library of the “Scikit-Learn library” contains the “train_test_split” method that allows us to seamlessly divide data into training and test sets with 80% Train dataset & 20% Test dataset.
# Splitting the human dataset into the training set and test set with (80% train & 20% test)
X_train, X_test, y_train, y_test = train_test_split(X_human,
y_human,
test_size = 0.20,
random_state=42)Choose a Model
In this project I will choose two machine learning algorithms:
1- Support Vector Machine algorithm (SVM): is a supervised machine learning algorithm that can be used for both classification or
regression challenges. However, it is mostly used in classification problems. In the SVM algorithm, we plot each data item as a point
in n-dimensional space (where n is the number of features you have) with the value of each feature being the value of a particular
coordinate. Then, we perform classification by finding the hyper-plane that differentiates the two classes very well
(look at the below snapshot).

2- K-Nearest Neighbors Algorithm (KNN): can be used for both classification and regression predictive problems. However, it is more
widely used in classification problems in the industry.
To evaluate any technique we generally look at 3 important aspects:
1- Ease to interpret the output
2- Calculation time
3- Predictive Power
Let’s take a simple case to understand this algorithm. Following is a spread of red circles (RC) and green squares (GS), You intend to find out the class of the blue star (BS). BS can either be RC or GS and nothing else. The “K” is KNN algorithm is the nearest neighbor we wish to take the vote from. Let’s say K = 3. Hence, we will now make a circle with BS as the center just as big as to enclose only three data points on the plane. Refer to the following diagram for more details:
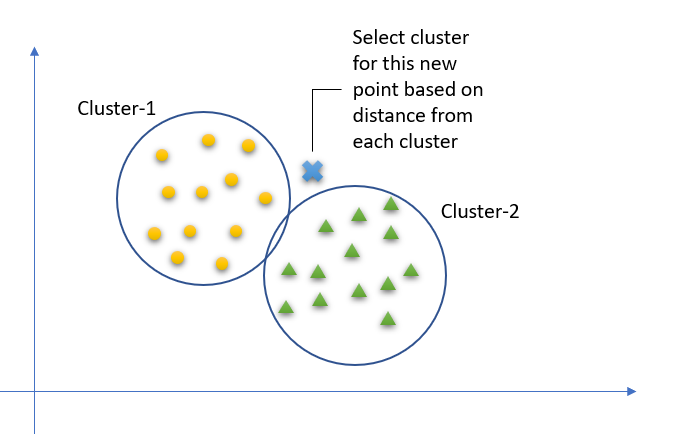
The three closest points to BS are all RC. Hence, with a good confidence level, we can say that the BS should belong to the class RC.
Here, the choice became very obvious as all three votes from the closest neighbor went to RC. The choice of the parameter K is very
crucial in this algorithm.
Train the Model
1- Training Support Vector Machine algorithm (SVM): We have divided the data into training and testing sets.
Now is the time to train our SVM on the training data. Scikit-Learn contains the SVM library, which contains built-in classes for different
SVM algorithms. Since we are going to perform a classification task, we will use the support vector classifier class, which is written as
SVC in the Scikit-Learns’ SVM library. This class takes one parameter, which is the kernel type. This is very important. In the case of
a simple SVM, we simply set this parameter as “linear” since simple SVMs can only classify linearly separable data.
#The fit method of SVC class is called to train the algorithm on the training data,
#which is passed as a parameter to the fit method
from sklearn.svm import SVC
svclassifier = SVC(kernel='linear')
svclassifier.fit(X_train, y_train)
2- Training K-Nearest Neighbors Algorithm (KNN): The first step is to import the “KNeighborsClassifier” class from
“sklearn.neighbors” library. In the second line, this class is initialized with one parameter, i.e. “n_neigbours”.
This is basically the value for the K. There is no ideal value for K and it is selected after testing and evaluation,
however, to start out, 5 seems to be the most commonly used value for the KNN algorithm.
#The fit method of KNN class is called to train the algorithm on the training data,
#which is passed as a parameter to the fit method
from sklearn.neighbors import KNeighborsClassifier
KNNclassifier = KNeighborsClassifier(n_neighbors=5)
KNNclassifier.fit(X_train, y_train)
Prediction
1- Making Prediction of SVM:
# enter all test datatest in model
y_pred = svclassifier.predict(X_test)2- Making Prediction of KNN:
# enter all test datatest in model
y_pred = KNNclassifier.predict(X_test) 6- Evaluate the Model:
Once you have built your model, the most important question that arises is
how good is your model?
Evaluating your model is the most important task in the data science project which delineates how good your predictions are.
The Confusion Metrics (Accuracy, Precision, Recall & F1 Score): is metrics using in evaluating the model by using (Accuracy, Precision, Recall, F1 score),

True Positives (TP) – These are the correctly predicted positive values which mean
that the value of the actual class is yes and the value of the predicted class is also yes.
True Negatives (TN) – These are the correctly predicted negative values which mean
that the value of the actual class is no and value of the predicted class is also no.
False Positives (FP) – When the actual class is no and the predicted class is yes.
False Negatives (FN) – When actual class is yes but predicted class is no.
1- Accuracy = TP+TN/TP+FP+FN+TN
2- Precision = TP/TP+FP
Precision – Precision is the ratio of correctly predicted positive observations to
the total predicted positive observations.
3- Recall = TP/TP+FN
Recall (Sensitivity) – Recall is the ratio of correctly predicted positive observations
to all observations in actual class – yes.
4- F1 Score = 2*(Recall * Precision) / (Recall + Precision)
F1 score – F1 Score is the weighted average of Precision and Recall.
1- Import SVM Confusion Metrics:
from sklearn.metrics import classification_report, confusion_matrix
print(confusion_matrix(y_test,y_pred))
print(classification_report(y_test,y_pred))
2- Import KNN Confusion Metrics:
from sklearn.metrics import classification_report, confusion_matrix
print(confusion_matrix(y_test,y_pred))
print(classification_report(y_test,y_pred))
